Aiming to enhance halide double perovskites technological applications, this research examines optoelectronic, structural, and mechanical properties of Rb2MgSnY6 (Y = I, Br, Cl) compounds via the first-principles method, evaluating their suitability for prospective applications. The optimised structural parameters and cell volumes expand proportionally with the size of the halogen atoms, and the computed tolerance factors, along with positive phonon frequencies in band structures, confirm both structural and dynamical stability. Electronic band structure analysis reveals that all examined compounds exhibit semiconducting characteristics, with a bandgap of 1.39, 1.95, and 2.45 eV, respectively, for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6. Mechanical analysis confirmed stability criteria and also demonstrated anisotropic and ductile behaviour. A range of optical parameters is analysed, such as dielectric function, absorption rate, optical response, and index of refraction for Rb2MgSnY6 (Y = I, Br, Cl) across the energy range of 0–40 eV. The results of the optical analysis reveal that these materials exhibit high optical conductivity, low reflectivity, and strong absorption ability. Overall, the structural, thermodynamic, and mechanical robustness emphasises the superb prospects of these compounds for deployment in solar cells, photodetectors, light-emitting diodes (LEDs), and various additional optoelectronic appliances.
Introduction
The energy demand is escalating gradually with the growing global population and is becoming a significant challenge to the scientific advancement and economic development of nations. Humans still rely heavily on conventional energy sources, such as oil, gas, and coal, to power most of the devices used in daily life. However, in recent times, it has become evident that these conventional energy resources will soon be in short supply if spent at the current rate and the world will ultimately face the challenge of mounting energy prices. In addition, the reliance on fossil fuels greatly contributes to greenhouse gas emissions, which in turn accelerates climate change. Alternatively, solar, wind, and geothermal energy are among the most significant non-conventional resources available in abundance in nature, but the absence of efficient energy storage systems limits their effective use. This realisation has motivated researchers to explore alternative energy sources that can serve humanity as effectively, or even better than conventional energy. It was soon recognised that solar energy and waste heat are vast sources of energy that can reduce dependence on traditional energy sources to build effective, sustainable, and eco-friendly energy conversion and storage systems. Alternative energy sources, such as sunlight and waste heat, are used in photovoltaic and thermoelectric applications, where the effectiveness of the application depends on the material selection. The materials choice plays a significant role, highlighting the need for efficient, stable and cost-effective solar cell materials and technologies to advance industries. In these applications, perovskite halides play a crucial role, which represents a significant breakthrough in materials research due to their versatility and strong potential in solar energy applications. They are typified via the generic formula ABX3 or A2BX6, identified as single or double perovskites. Unlike single perovskite, the structural flexibility of double perovskites accommodates cations with charges ranging from 1+ to 4+ at the B-site, where single perovskite is restricted to only 2+ cations [1]. Therefore, double perovskite exhibits enhanced perfor-mance, better adaptability, greater tunability, improved long-term stability, fewer defects, as well as greater flexibility in tailoring the bandgap.
Many materials with various structural types have been investigated for use in the manufacturing of optical devices. However, substances with perovskite-type configurations have garnered considerable attention due to the interesting characteristics these materials exhibit. Previously, various materials, such as Pb2NaIO6, Cs2PbX6 (where “X” repre-sents I, Br, Cl), and CsPbM3 (where “M” is Cl, I, and Br), have been studied and reported by researchers for use in optoelectronic devices. However, material engineers avoid these materials in the manufacturing of practical devices because of lead, a noxious element that exists in these materials [2–4]. Therefore, when investigating materials for optical devices, researchers should avoid those contain-ing toxic elements. Additionally, optical devices should be made from materials that are affordable to facilitate broader human use. In the past, many other double perov-skite halide materials (DPHM), for instance Cs2AuBiX6 (where X = Cl, Br), Rb2AgBiX6 (X = Br, I), Cs2AgBiCl6, Cs2AgInCl6, and Cs2AgBiI6 have been developed, but the presence of silver or gold makes them expensive materials for practical applications [5–8]. Recently, a research group investigated rubidium Rb-based double perovskites, i.e., Rb2XCl6 (where X = Se, Ti), and they found that these materials exhibit semiconducting properties with bandgaps of 2.95 eV and 2.84 eV, respectively, between the valence and conduction bands. Additionally, they determined that both materials are chemically and dynamically stable [9]. Manzoor et al. computationally studied the Rb2YAgBr6 and Rb2YAgI6 compounds and found that they have indirect bandgap natures of 4.2 and 3.2 eV, respectively [10]. Although these materials were found to have a semicon-ducting nature, their wide bandgaps limit their applicability in a wide range of applications. Similarly, many other research groups have also studied Rb-based double perov-skites via the density functional theory (DFT) approach [11–13].
In this context, numerous researchers have devoted their efforts to significant theoretical studies on double halide perovskites. Conspicuously, Haq and his coworkers investigated the thermal and opto-electronic characteristics of Rb2XGaBr6 (X = K, Na) compounds and found that their bandgaps were 1.90 and 2.2 eV, respectively [14]. McClure and his team found that the indirect wide bandgap of Cs2AgBiCl6 and Cs2AgBiBr6 exceeds the optimal range for solar cells, potentially reducing their efficiency by up to 2.5% [15]. Aldaghfag et al. found that K2ScAgCl6 and Na2ScAgCl6 are direct bandgaps materials of 3.65 and 3.63 eV, highlighting their potential for use in UV photodetectors and sunlight absorbing applications [16]. Mahmud and his team recently examined A2AuScX6 (A = Cs, Rb and X = Cl, Br, I) DPHM and found them to exhibit favourable visible light bandgaps between 1 to 2 eV [17]. The previously mentioned studies have confirmed extensive research on double halide perovskites. However, to date, neither experimental nor theoretical efforts have explored the properties of Rb2MgSnY6 (Y = I, Br, Cl) compounds. Hence, inspired by this research gap, we are drivien to examine the potential of Rb-based halide perovskites, specifically Rb2MgSnY6 (Y = I, Br, Cl), in photovoltaic and optoelectronic devices. In this research, a comprehensive theoretical evaluation of the optoelectronic, structural, and mechanical characteristics is presented, employing the first-principles method to elucidate the fundamental characteristics of these materials.
Computational model
An in-depth analysis conducted with the WIEN2k compu-tational package [18] explored the structural, optical, electronic, and mechanical characteristics of inorganic double halide perovskites Rb2MgSnY6 (Y = I, Br, Cl) via the first-principles technique. The calculations were performed using the generalised gradient approximation (GGA), as formulated by Perdew, Burke, and Ernzerhof (PBE) [19]. While the GGA approach typically diminishes the energy gap of the material, Tran and Blaha modified the Becke-Johnson (TB-mBJ) potential [20], which was also employed for enhancing accuracy in calculating the band gap. The Kohn–Sham equation was solved using the full potential linearised augmented plane wave (FP-LAPW) [21] approach in combination with local orbitals, while examining the diverse characteristics of the titled com-pounds. An appropriate muffin-tin radius (RMT) values are selected to ensure convergence of total energy to prevent any loss of electronic charge from within the core region of atoms. A value for RKmax is chosen as 7, where R represents the muffin-tin radius to maintain calculation accuracy, and Kmax denotes the cutoff wave vector in the expansion of the plane wave [22]. Meanwhile, the cutoff energy, which distinguishes the energy gap between the core and valence bands, is set to −6.0 Ry. The total energy-volumetric data were used to establish the lattice parameters through the Birch–Murnaghan state equation fitting [23]. Functions of spherical harmonics are used within the muffin-tin sphere, with a cutoff of maximum angular momentum (Lmax) set to 10 and a maximum Fourier expansion coefficient of charge density (Gmax) set to a value of 12. Successively, to refine the lattice parameters, this value was subsequently abridged by 3.5%. The convergence criterion for energy and charge at 0.001 Ry, along with 0.00001 e, correspondingly was set in self-consistent field (SCF) computations. This calculation relied on the Monkhorst–Pack [24] k-point mesh design, with a 10 × 10 × 10 k-mesh to address the Brillouin zone using 2000 k-points. The IRelast package [25] can be used to determine the mechanical character-istics of titled compounds, for instance, their elastic constants, which provide insight into their stiffness and structural stability. For the computation of phonon disper-sion, the CASTEP code [26, 27] is used, which is essential for understanding the thermodynamic behaviour of titled compounds.
Results and discussions
Structural properties
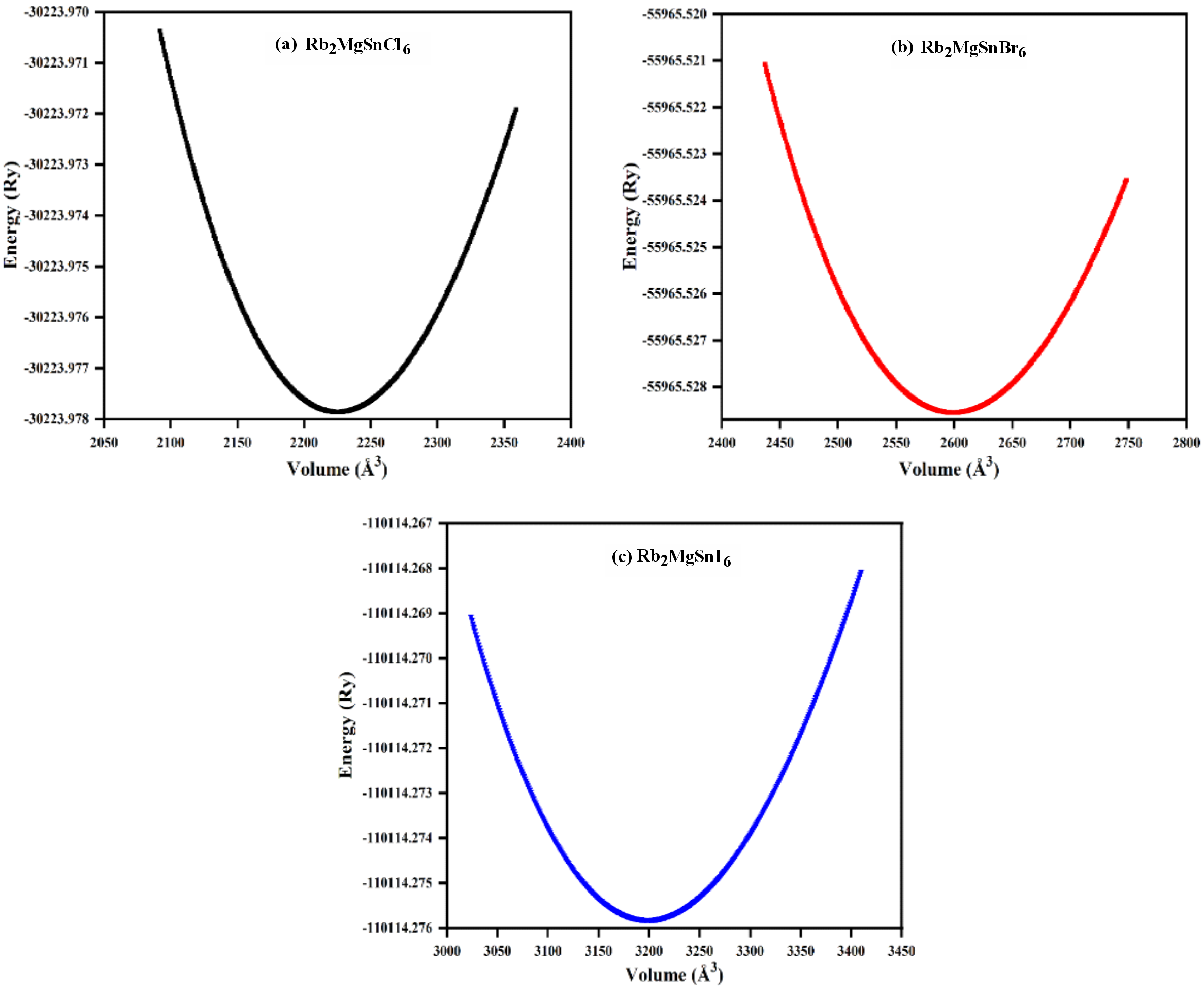
Analysing solid state materials requires structural properties as a key factor for understanding their various charac-teristics. This work analysed the structural features of Rb2MgSnY6 (where Y = I, Br, Cl) DPHM and their atoms were positioned as illustrated in Fig. 1. First, lattice parameters of all the titled compounds were evaluated from the respective optimised volume vs. energy curve and depicted in Fig. 2 with corresponding values listed in Table 1. Among the studied titled compounds, the material having chlorine as the non-metal at the “Y” place showed the lowest lattice constant. However, the lattice constants of the Rb2MgSnY6 (where Y = Br, I) were higher than those of the Rb2MgSnCl6 compound, and th(is increment was due to having higher ionic radii of the bromine and iodine than that of the chlorine atom [2]. Moreover, from their respective optimised energy-volume curves, the ground state energies, ground state volumes, and bulk modulus were determined, and the results are summarised in Table 1. From Table 1, it is evident that these parameters are also dependent on the non-metal replacement in the Rb2MgSnY6 (where Y = I, Br, Cl) DPHM. During the study, it was found that Rb2MgSnl6 has the highest value of volume, while its bulk modulus and energy values were the lowest among the studied Rb2MgSnY6 (Y = I, Br, Cl) compounds.
Calculated structural parameters for Rb2MgSnY6 (Y = I, Br, Cl) compounds
Using (1), the tolerance factor (τ) values of all three studied DPHM were determined, and it was found that their values lie between 0.8 and 1, as suggested by Bartel et al. for stable materials [28]. Lastly, for the experimental synthesis of the studied Rb2MgSnY6 (Y = I, Br, Cl) compounds, their formation energies (Ef) were calculated using (2). All the currently studied materials exhibited higher negative values of Ef, which validates the possibility of their experimental formation [29]:
where in (1), rA, rB, and rX represent the ionic radii of A‑site, B-site (i.e., B + B′/2), and X-site, respectively, in the general A2BB′X6 formula. While in (2), ERb2MgSnY6 signifies the total energy of the Rb2MgSnY6 (Y = I, Br, Cl) com-pounds, and ERb, ESn, and EY represent the individual energies of Rb, Mg, Sn, and Y (Cl, Br, I) atoms, respectively.
Phonon dispersion analysis was also conducted for the studied Rb2MgSnY6 (Y = I, Br, Cl) double halide perovskites to check the dynamical stabilities of these compounds, and their final patterns are illustrated in Fig. 3. From their phonon patterns, it is obvious that these materials possess dynamic stabilities due to absence of imaginary frequencies in their plots, exhibiting a similar trend to that reported in the previous study by Sajjad et al. [30]. The phonon results confirm the structural strength of the titled compounds, indicating that these materials are suitable for practical applications.
Furthermore, thermodynamic stabilities of the Rb2MgSnY6 were also investigated via the ab initio molecu-lar dynamics (AIMD) method at room temperature [31]. For thermodynamic stabilities of the given substances, we monitored temperature and total energy deviations with time which is presented in Fig. 4. Both energy and tem-perature curves of all the given substances showed minor variations throughout the range, ensuring their stabilities at 300 K. Additionally, during the AIMD calculations, no new phases or breakage of bonds were detected in the given substances, authenticating structural integrity of titled compounds at room temperature.
Fig. 4.The energy and temperature plots as a function of simulation time for (a) Rb2MgSnCl6, (b) Rb2MgSnBr6,
(c) Rb2MgSnl6 compounds.
Electronic properties
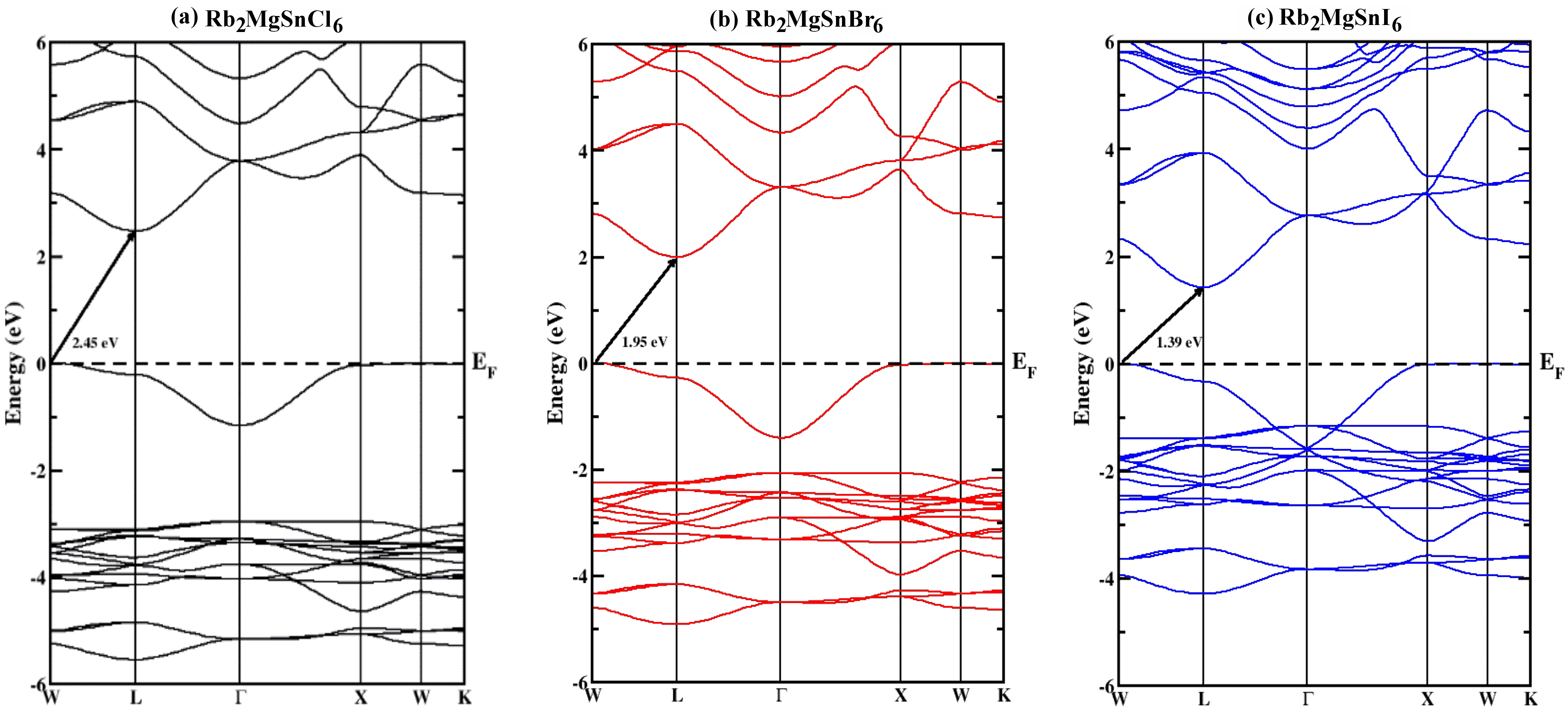
The electronic properties of all the Rb2MgSnY6 (Y = I, Br, Cl) compounds were elaborated to understand the behaviour of these materials. Significant insights into their electronic properties were obtained, providing valuable information. We depicted the band structure calculation along a higher symmetric direction in Fig. 5. From Fig. 5, it is apparent that in all the materials, the valence band curves touch the Fermi line but do not cross it. Moreover, there is a clear bandgap between the conduction and valence bands in all studied materials. This behaviour reinforces the semi-conducting character of the given substances. Fig. 5 also shows that the gaps between the valence and conduction bands vary depending on the non-metal replacement at the “Y” position in the studied Rb2MgSnY6 (Y = I, Br, Cl) substances. The bandgap decreased as chlorine was replaced with bromine and iodine at the “Y” position. This is because the bandgaps are impressively influenced by lattice parameters and electronegativity [4]. All three compounds showed an indirect nature of bandgap from W-L momentum points, as indicated in Fig. 5 by an arrow symbol. The noted values for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6 bandgaps are 1.39, 1.95, and 2.45 eV, respectively. The bandgaps calculated for the studied compounds are highly favourable, making these materials excellent candidates for solar cells, photodetectors, LEDs, and additional optoelectronic appliances [2, 32].
Fig. 5.Calculated band structures of (a) Rb2MgSnCl6 (b) Rb2MgSnBr6, (c) Rb2MgSnl6 compounds.
The titled materials electronic spectra were further elaborated by calculating the total density of states (TDOS) for each of the studied compounds. Their results are presented in Fig. 6. It is apparent from Fig. 6 that the valence band curves of all studied compounds do not traverse the Fermi level. This behaviour of the studied materials authenticates the band structure results of these substances. Also, the partial density of states (PDOS) of each studied compound was calculated, and their findings are illustrated in Fig. 6. This provides information about the contribution of each atom within the compound. From the PDOS of the studied compounds, it is clear that in the valence bands of the studied materials, the major peaks were seen for the non-metals placed at the “Y” position in Rb2MgSnY6 (Y = I, Br, Cl), while the other atoms have small contributions. The major peaks in the conduction band were detected for the “Sn” and non-metal atoms, while “Rb” and “Mg” showed minimal contributions.
Fig. 6.TDOS and PDOS patterns of the (a) Rb2MgSnCl6 (b) Rb2MgSnBr6, (c) Rb2MgSnl6 substances.
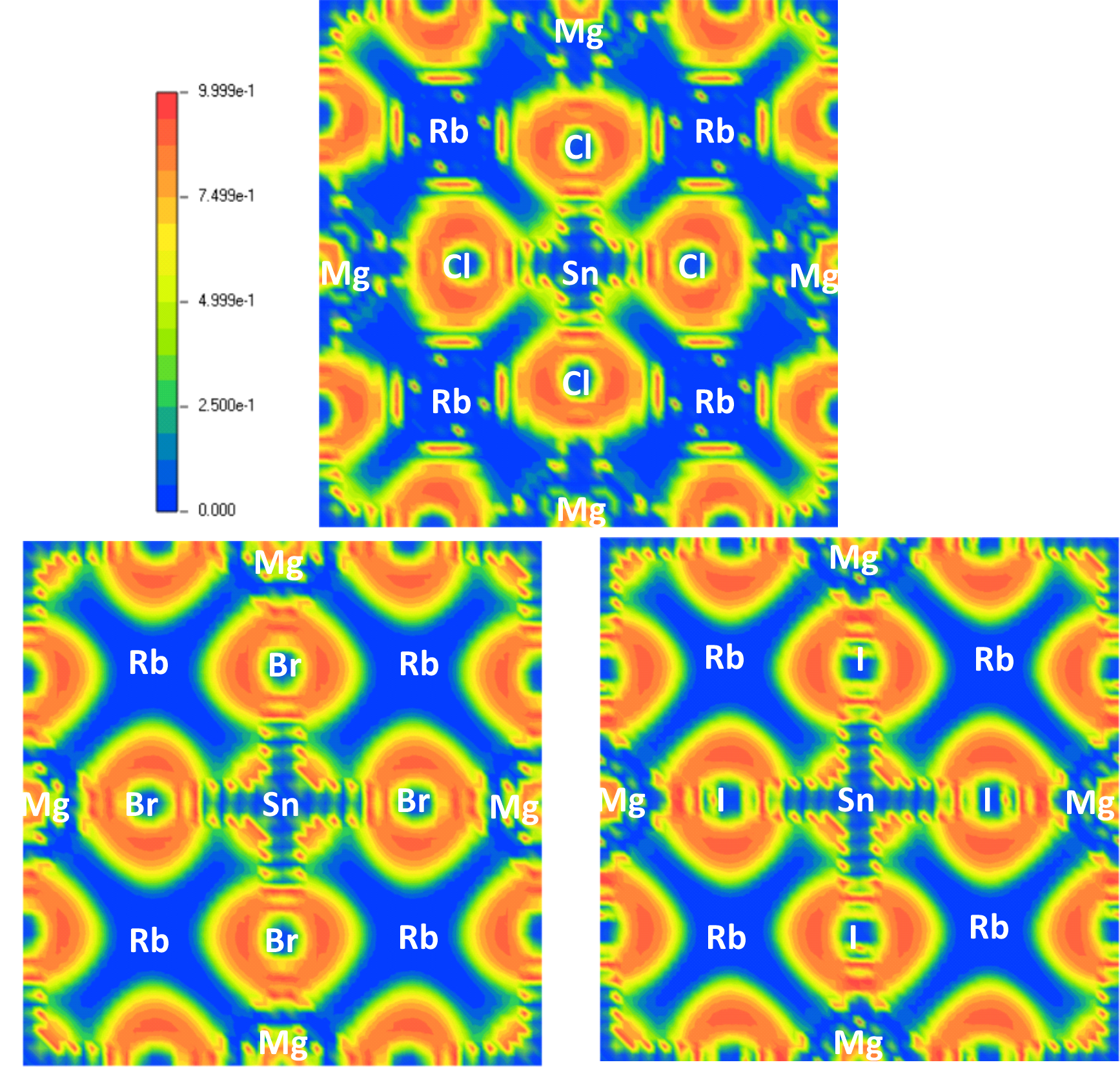
The electron localisation function (ELF) maps for Rb2MgSnY6 (Y = Cl, Br, I) perovskites provide insight into the electron density distribution within these structures. The ELF analysis presented in Fig. 7 shows the prominent electron localisation around the non-metal atoms (Cl, Br, I). This distribution suggests that electron density is transported from electronegative elements in the first column of the periodic table, such as Rb, which exhibits minimal electron density, towards the highly electronegative halogens [33]. Atoms of Sn, possessing transitional electro-negativity, exhibit reasonable electron density, reflecting partial electron distribution within bonds of Sn-X. Additionally, the nature of bonding is examined using charge density and spatial distribution visualisations. In contrast to covalent bonds, where electrons are shared, ionic bonds are marked by localised electron density around distinct atoms, typically near 1 eV. The distribution plots highlight this distinction, showcasing regions of shared electron density between halogens and Sn or Mg atoms, emphasising the covalent nature of these interactions. Among the studied compounds, Rb2MgSnBr6 exhibits the strongest covalent bonding character in its Sn-Br and Mg-Br bonds, reflecting its unique electronic structure.
Fig. 7.Electron localisation function (ELF) of Rb2MgSnY6.
Mechanical properties
Before using any material in practical applications, it is necessary to understand its mechanical behaviour. This property of any material provides information about the overall behaviour of that material under the implementation of any external stress. To evaluate the mechanical properties of a material, the main task is to determine its three main elastic (C11, C12, and C44) constants. Through these elastic constants, additional mechanical characteristics of the materials under consideration can be further determined. For the currently studied Rb2MgSnY6 (Y = I, Br, Cl) DPHM, these constants were computed, and the results are tabulated in Table 2. Subsequently, using the equations {i.e., (3), (4), (5), (6), (7), and (8)}, all other mechanical parameters were calculated for the given substances, with corresponding values listed in Table 2. Table 2 indicates that the calculated elastic constants for the studied materials satisfy the basic stability conditions (for instance, C11 + 2C12 > 0, C11 and C44 > 0, and C11 – C12 > 0) for cubic material, consistent with the conditions reported by Rahman et al. and other previous studies [34–37].
Calculated mechanical parameters for Rb2MgSnY6 (Y = I, Br, Cl) double perovskites
From Table 2, it is clear that Rb2MgSnCl6 has higher values of bulk modulus (B), shear modulus (G), and Young’s modulus (E), indicating that this material has a higher tendency to keep its shape, length, and volume under the implementation of external stress than the other two studied materials. It has been previously reported by Israr et al. and other studies that a material with isotropic properties must have an anisotropy factor (A) value of precisely 1, while materials with A values either higher or less than 1 exhibit anisotropy properties [38, 39]. Since none of the studied titled substances has an A value of exactly 1, it means that all these materials are anisotropic crystals. Furthermore, based on the values of Poisson’s ratio (V) and Pugh’s ratio (B/G) calculated for all these studied materials, it was found that they all exhibit a ductile nature. This conclusion is supported by their B/G values being higher than 1.75 and none of the V values being less than 0.26 [40, 41].
The essential optical parameters that need to be elaborated for semiconducting materials are optical conductivities σ(ω), complex dielectric functions ε(ω), absorption coeffi-cients α(ω), reflectivity R(ω), and refractive index η(ω). The dielectric function shows the overall disturbance caused by the interaction of electromagnetic radiation with the materials, and it can be described entirely by the following equation [42]:
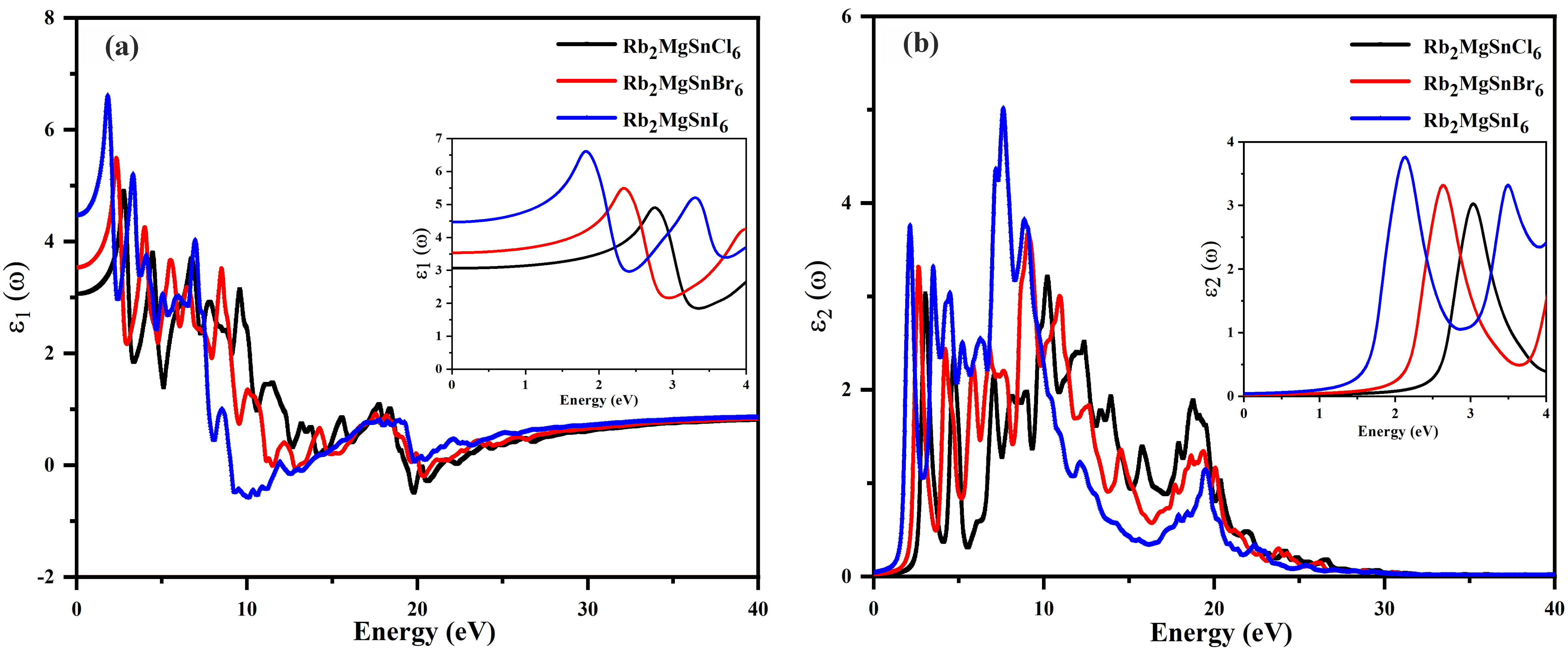
where ε1(ω) and ε2(ω) correspond to the real and imaginary parts of the dielectric function. For Rb2MgSnY6 DPHM, these real and imaginary parts were calculated and illustrated in Fig. 8(a) and Fig. 8(b), respectively. The ε1(ω) provides overall information about the polarisation and dispersion of electromagnetic photons from the material lattice. From Fig. 8(a), it is obvious that the static values are 3.1, 3.6, and 4.4 for Rb2MgSnCl6, Rb2MgSnBr6, and Rb2MgSnl6, respectively. Fig. 8(a) also shows that the highest peaks are 6.6 at 1.8 eV, 5.5 at 2.32 eV, and 4.98 at 2.48 eV for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6, respectively. Moreover, as the photon energy, all the titled compounds exhibited additional peaks at slightly lower values than the initially observed peaks at 1.8 eV, 2.32 eV, and 2.48 eV, respectively. However, it was found that Rb2MgSnl6 at 9.1 eV, Rb2MgSnBr6 at 11.7 eV, and Rb2MgSnCl6 at 19.6 eV crossed the zero level, demonstrating their metallic nature, which authenticates that at this level, photons cannot penetrate the material [43, 44]. The imaginary part ε2(ω) represents the transition of an electron from the valence to the conduction band [45] as depicted in Fig. 8(b). Fig. 8(b) represents the imaginary dielectric constant data for all the given substances. These data provide information about the absorption of incident photons by the materials. From Fig. 8(b), it is clear that at 1.39 eV, 1.95 eV, and 2.45 eV, the given substances reach their threshold absorption values. Later, with increasing photon energy, all materials exhibited a fluctuating behaviour in their imaginary values. At photon energies higher than 30 eV, all three materials showed no significant increase in their imaginary dielectric constant values.
Fig. 8.Dielectric function of (a) real parts (b) imaginary parts of the Rb2MgSnY6 compounds.
The refractive index of Rb2MgSnY6 (Y = I, Br, Cl) substances follows the same trend of variation as ε1(ω) with increasing photon energy, and both are related mathemati-cally by the following equation:
\(
\eta^2-k^2=\varepsilon_1(\omega)
\) (10)
For any material, it is necessary to satisfy the relation η02 = ε1(0). From Fig. 8(a) and Fig. 9(a), it is clear that the currently studied substances satisfy this relation [45]. It has been reported that materials with higher refractive index values are more suitable for applications operating in the visible range [44]. From Fig. 9(a), it is evident that all the studied materials can operate within the visible photon energy range. Moreover, the given substances exhibit additional peaks at higher photon energies, indicating that they can also operate efficiently in the ultraviolet region. Fig. 9(b) shows the absorption curves of Rb2MgSnY6 (Y = I, Br, Cl) DPHM. The following equation can mathematically express the absorption coefficient of any material:
Fig. 9(b) shows the ability of titled compounds in the 0–40 energy range of photons. It is clear from Fig. 9(b) that Rb2MgSnl6 material showed an initial response of absorption to the incident photons of lower energy due to its lower bandgap compared to the other two studied materials. Initial peaks of absorption were detected as 18.8 × 104/cm at 2.2 eV, 22.6 × 104/cm at 2.7 eV, and 25.1 × 104/cm at 3.1 eV for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6, respectively, within the visible (1.6 to 3.26 eV) energy range. The Rb2MgSnCl6 shows a greater tendency for absorption in the visible energy range. Moreover, in the ultraviolet energy range, the highest values found for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6 DPHM were 132.6 × 104/cm at 19.7 eV, 161.9 × 104/cm at 19.9 eV, and 193.9 × 104/cm at 19.6 eV, respectively. From the calculated absorption values of the given materials in both the visible and ultraviolet energy ranges, it is clear that Rb2MgSnCl6 has the greatest ability to absorb in both ranges.
The optical conductivities of the studied materials are presented in Fig. 9(c). All the titled materials show their optical conductivity in both the visible and ultraviolet energy regions. It has been reported that materials should have good optical conductivity in the energy range of 1–3.5 eV to be fit for the manufacturing of solar cells [2]. From Fig. 9(c), it is clear that all materials have shown good optical conductivity within the required energy range for solar cell applications. The highest optical conductivity values noted for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6 in the visible energy range were 1083 (Ω · cm)-1 at 2.13 eV, 1181 (Ω · cm)-1 at 2.65 eV, and 1236 (Ω · cm)-1 at 3.03 eV, respectively. Additionally, in the ultraviolet energy region, the given compounds present much higher values of optical conductivity. The calculated optical conductivities for Rb2MgSnl6, Rb2MgSnCl6, and Rb2MgSnBr6 were 5134 (Ω · cm)-1 at 7.63 eV, 4764 (Ω · cm)-1 at 18.76 eV, and 4556 (Ω · cm)-1 at 8.85 eV, respectively. However, with increasing photon energy in the ultraviolet energy range beyond 20 eV, no specific increase in optical conductivity of the given substances was observed. Based on these results of optical conductivity, the titled compounds show the potential for use in solar cells and other optoelectronic devices.
Fig. 9(d) represents the reflection of photons from the surfaces of Rb2MgSnY6 (Y = I, Br, Cl) DPHM in the energy range of 0–40 eV. The highest values of reflectivity found for Rb2MgSnl6, Rb2MgSnCl6, and Rb2MgSnBr6 DPHM were 0.21 at 2 eV, 0.18 at 2.51 eV, and 0.16 at 3.25 eV, respectively, in the visible energy range. However, these reflectivity values were found to be slightly higher in the ultraviolet energy range than in the visible energy range. The calculated reflectivity values for Rb2MgSnl6, Rb2MgSnCl6, and Rb2MgSnBr6 were found to be 0.28 at 9.21 eV, 0.30 at 19.85 eV, and 0.25 at 20.47 eV, respectively. Overall, the proportion of reflected photons from the surfaces of Rb2MgSnY6 in both the visible and ultraviolet energy regions is quite low, which further enhances their suitability for practical applications.
Therefore, from the overall study of the optical characteristics of Rb2MgSnY6 compounds, it is clear that these materials exhibit high optical conductivity, very low reflectivity, and strong absorption ability in both visible and ultraviolet energy ranges. Consequently, these materials are best for applications of solar cells, photodetectors, LEDs, and other optoelectronics devices.
Conclusions
Theoretical calculations for the presently studied Rb2MgSnY6 (Y = I, Br, Cl) DPHM were performed using the FP-LAPW technique. The computed tolerance factor (τ) values of the given materials fall between 0.8 and 1, as suggested by Bartel et al. for stable perovskites. Phonon dispersion and AMID results ensure the dynamical and thermal stabilities of all the given substances, respectively. Their formation energies were calculated, and it was observed that all the studied compounds have highly negative formation energies, which enhances their practical synthesisability. Analysis of electronic spectra shows an indirect bandgap nature at W-L symmetry points, and the noted values of bandgaps for Rb2MgSnl6, Rb2MgSnBr6, and Rb2MgSnCl6 were 1.39 eV, 1.95 eV, and 2.45 eV, respectively. Mechanical analysis confirms that all the investigated compounds display higher computed values of B/G and V than 1.75 and 0.26, respectively, confirming their ductile behaviour. Moreover, the calculated elastic constants further validate their stability and anisotropy behaviour. Multiple optical parameters are analysed, including dielectric functions, absorption coefficients, optical conductivity, refractive index and related features. The results suggest that all three compounds display strong absorption, high conduction, and low reflectivity. Based on the results of the optical conductivities in both in the visible and ultraviolet energy ranges, these materials are strong contenders for use of optoelectronic technologies.
Competing interests
The authors declare no relevant financial or non-financial interests.
References
Dar, S. A. & Want, B. Direct band gap double perovskite halide Cs2ScInCl6 for optoelectronic applications – A first principle study. Comput. Condens. Matter 33, e00736 (2022). https://doi.org/10.1016/j.cocom.2022.e00736
Hussain, M., Rashid, M., Ali, A., Bhopal, M. F. & Bhatti, A. S. Systematic study of optoelectronic and transport properties of cesium lead halide (Cs2PbX6; X = Cl, Br, I) double perovskite for solar cell applications. Ceram. Int. 46, 21378–21387 (2020). https://doi.org/10.1016/j.ceramint.2020.05.235
Aziz, A. et al. Theoretical investigation of X2NaIO6 (X = Pb, Sr) double perovskitefor thermoelectric and optoelectronic applications. Phys. B: Condens. Matter 630, 413694 (2022). https://doi.org/10.1016/j.physb.2022.413694
Murtaza, G. & Ahmad, I. First principle study of the structural and optoelectronic properties of cubic perovskites CsPbM3 (M = Cl, Br, I). Phys. B: Condens. Matter 406, 3222–3229 (2011). https://doi.org/10.1016/j.physb.2011.05.028
Mathew, N. P., Kumar, R. & Radhakrishnan, R. First principle study of lead free halide double perovskite Cs2AuBiX6 (X = Cl, Br). Mater. Today: Proc. 27, 561–564 (2020). https://doi.org/10.1016/j.matpr.2019.12.022
Mehed, A. M., Al-Qaisi, S. & Ali, M. A. Study of optoelectronic and thermoelectric properties of double perovskite Rb2AgBiX6, (X = Br, I): by DFT approach. Eur. Phys. J. Plus 137, 990 (2022). https://doi.org/10.1140/epjp/s13360-022-03222-4
Alotaibi, N. H. et al. DFT study of double perovskites Cs2AgBiX6 (X = Cl, Br): An alternative of hybrid perovskites. J. Solid State Chem. 313, 123353 (2022). https://doi.org/10.1016/j.jssc.2022.123353
Mathew, N. P., Kumar, N. R. & Radhakrishnan, R. First principle study of the structural and optoelectronic properties of direct bandgap double perovskite Cs2AgInCl6. Mater. Today: Proc. 33, 1252–1256 (2020). https://doi.org/10.1016/j.matpr.2020.03.489
Al-Qaisi, S. et al. A theoretical investigation of the lead-free double perovskite halides Rb2XCl6 (X = Se, Ti) for optoelectronic and thermoelectric applications. J. Comput. Chem. 44, 1690–1703 (2023). https://doi.org/10.1002/jcc.27119
Manzoor, M. et al. DFT study of electronic, optical, and elastic properties of double perovskites Rb2YAgX6 (X = Br, I) compounds for optoelectronic device applications. Phys. Scr. 98, 035703 (2023). https://doi.org/10.1088/1402-4896/acae0e
Yaseen, M., Aldaghfag, S. A., Zahid, M. & Misbah. Physical characteristics of X2NaMoBr6 (X = Rb, K): A DFT study. Mater. Sci. Semicond. Process. 147, 106760 (2022). https://doi.org/10.1016/j.mssp.2022.106760
Albalawi, H. et al. Study of new double perovskite halides Rb2Ti (Cl/Br)6 for solar cells and thermoelectric applications. Mater. Today Commun. 32, 104106 (2022). https://doi.org/10.1016/j.mtcomm.2022.104106
Manzoor, M. et al. Probing direct bandgap of double perovskite Rb2LiTlX6 (X = Cl, Br) and optoelectronic characteristics for solar cell applications: DFT calculations. J. Mater. Res. Technol. 18, 4775–4785 (2022). https://doi.org/10.1016/j.jmrt.2022.04.073
Haq, A. U., Mustafa, G. M., Amin, M., Ramay, S. M. & Mahmood, A. Ab-initio study of opto-electronic and thermoelectric properties of direct bandgap double perovskites Rb2XGaBr6 (X = Na, K). Int. J. Energy Res. 45, 9241–9251 (2021).https://doi.org/10.1002/er.6455
McClure, E. T., Ball, M. R., Windl, W. & Woodward, P. M. Cs2AgBiX6 (X = Br, Cl): New visible light absorbing, lead-free halide perovskite semiconductors. Chem. Mater. 28, 1348–1354 (2016). https://doi.org/10.1021/acs.chemmater.5b04231
Aldaghfag, S. A. et al. Investigation of electronic, optical and thermoelectric features of X2ScAgCl6 (X = K, Na) double perovskites for renewable energy applications. J. Solid State Chem. 312, 123179 (2022). https://doi.org/10.1016/j.jssc.2022.123179
Mahmud, S., Ali, M. A., Hossain, M. M. & Uddin, M. M. DFT aided prediction of phase stability, optoelectronic and thermoelectric properties of A2AuScX6 (A = Cs, Rb; X= Cl, Br, I) double perovskites for energy harvesting technology. Vacuum 221, 112926 (2024). https://doi.org/10.1016/j.vacuum.2023.112926
Blaha, P. et al. WIEN2k: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties. (Techn. Universitat, 2019).
Tran, F. & Blaha, P. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009).
Usman,T. et al. First-principles study of structural, electronic, optical and thermoelectric properties of rare earth based perovskites XAlO3 (X = Sm, Eu, Gd). Comp. Theor. Chem. 1235, 114567 (2024). https://doi.org/10.1016/j.comptc.2024.114567
Segall, M. D. et al. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 14, 2717–2744 (2002). https://doi.org/10.1088/0953-8984/14/11/301
Bartel, C. J. et al. New tolerance factor to predict the stability of perovskite oxides andhalides. Sci. Adv. 5, eaav0693 (2019). https://doi.org/10.1126/sciadv.aav0693
Al-Humaidi, J. Y. et al. First-principle insight into the structural, electronic, elastic and optical properties of Cs-based double perovskites Cs2XCrCl6 (X = K, Na). RSC Adv. 13, 20966–20974 (2023). https://doi.org/10.1039/D3RA03706A
Sajjad, A. et al. Exploring double perovskites Cs2AgSbX6 (X = Cl, Br, and I) as promising optoelectronic and thermoelectric materials: a first-principle study. Phys. Chem. Chem. Phys. 27, 4880–4891 (2025). https://doi.org/10.1039/D4CP04662E
Kraska, T. Stability limits of pure substances: An investigation based on equations of state. Ind. Eng. Chem. Res. 43, 6213–6221 (2004). https://doi.org/10.1021/ie049720v
Nakajima, T. & Sawada, K. Discovery of Pb-free perovskite solar cells via high throughput simulation on the K computer. J. Phys. Chem. Lett. 8, 4826–4831 (2017). https://doi.org/10.1021/acs.jpclett.7b02203
Archi, M. et al. The effect of doping/dual-doping with nitrogen and silicon on the structural, electronic, and optical properties of graphene: First-principles study. J. Nanopart. Res. 26, 138 (2024). https://doi.org/10.1007/s11051-024-06056-6
Afaq, A., Bakar, A., Anwar, S., Anwar, W. & Aleem, F. E. DFT study of SmXO3 (X = Al and Co) for elastic, mechanical and optical properties. Int. J. Mod. Phys. B 32, 1850362 (2018). https://doi.org/10.1142/S0217979218503629
Usman, T., Murtaza, G., Luo, H. & Mahmood, A. GGA and GGA+U study of rare Earth-based perovskites in cubic phase. J. Supercond. Nov. Magn. 30, 1389–1396 (2017). https://doi.org/10.1007/s10948-016-3953-9
Ahmed, M., Bakar, A., Quader, A., Ahmad, R. A. & Ramay, S. M. First-principles calculations to investigate structural, elastic, mechanical, electronic and optical characteristics of RbSrX3 (X = Cl,Br). Chem. Phys. 581, 112260 (2024). https://doi.org/10.1016/j.chemphys.2024.112260
Rahman, N. et al. Probing the physical properties of M2LiCeF6 (M = Rb and Cs) double perovskite compounds for prospective high-energy applications employing the DFT framework. RSC Adv.13, 15457–15466 (2023). https://doi.org/10.1039/D3RA01451G
Israr, N. et al. Exploring the structural, mechanical, and optical properties of K2InGaX6 (X = Cl, Br or I) compounds be density functional theory. J. Inorg. Organomet. Polym. Mater. (2025). https://doi.org/10.1007/s10904-025-03731-6
Yang, Y., Lu, H., Yu, C. & Chen, J. M. First-principles calculations of mechanical properties of TiC and TiN. J. Alloys Compd. 485, 542−547 (2009). https://doi.org/10.1016/j.jallcom.2009.06.023
Pugh, S. F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Phil. Mag. 45, 823–843 (1954). https://dx.doi.org/10.1080/14786440808520496
Noor, N. A. et al. Analysis of direct band gap A2ScInI6 (A = Rb, Cs) double perovskite halides using DFT approach for renewable energy devices J. Mater. Res. Technol. 13, 2491–2500 (2021). https://doi.org/10.1016/j.jmrt.2021.05.080
Shakeel, S., Song, P., Alsalmah, H. A., Murtaza, G. & Huang, T. Investigation of pressure-dependent electronic and optical properties of double perovskites Cs2AgXY6 (X = Bi, In; Y= Cl, Br). J. Inorg. Organomet. Polym. Mater. 34, 1040–1054 (2024). https://doi.org/10.1007/s10904-023-02888-2
Khan, N. et al. Detail computational study about the structural, electronic, optical and mechanical properties of RbVX3 (Cl, Br, I) halide perovskite materials. RSC Adv. 13, 22958–22965 (2023). https://doi.org/10.1039/D3RA03615D
Cahill, D. G., Watson, K. & Pohl, R. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 46, 6131 (1992). https://doi.org/10.1103/PhysRevB.46.6131
Shah, S. H. et al. uniaxial strain engineering of electronic, elastic and optical properties of halide double perovskites K2NaTIX6 (X = I, Br, and Cl): A DFT insight. J. Inorg. Organomet. Polym. Mater. 35, 3665–3681 (2025). https://doi.org/10.1007/s10904-024-03484-8